Practice Essentials
Hyperaldosteronism is characterized by excessive secretion of aldosterone, which causes increases in sodium reabsorption and loss of potassium and hydrogen ions. It may be either primary (autonomous) or secondary. [1, 2] Hyperaldosteronism represents part of a larger entity of hypermineralocorticoidism that may be caused by aldosterone, its mineralocorticoid precursors, or defects that modulate aldosterone effects on its target tissues. Surgical excision of the affected adrenal gland is recommended for all patients with hyperaldosteronism who have a proven aldosterone-producing adenoma (APA).
Aldosterone is a steroid hormone produced exclusively in the zona glomerulosa of the adrenal cortex. It is the major circulating mineralocorticoid in humans. Numerous aldosterone precursors, including deoxycorticosterone and 18-hydroxycorticosterone, have mineralocorticoid activity and may produce or exacerbate features typical of mineralocorticoid hypertension when present in excessive amounts in various pathologic states.
The principal site of action of aldosterone is the distal nephron, though several other sites of aldosterone-sensitive sodium regulation are noted, including the sweat glands and the gastrointestinal (GI) tract. The principal regulators of aldosterone synthesis and secretion are the renin-angiotensin system and the potassium ion concentration. Minor regulators include adrenocorticotropic hormone (ACTH) from the pituitary, atrial natriuretic peptide from the heart, and local adrenal secretion of dopamine.
Signs and symptoms of hyperaldosteronism
Primary hyperaldosteronism may be asymptomatic, particularly in its early stages. When symptoms are present, they may be related to hypertension (if severe), hypokalemia, or both.
The spectrum of hypertension-related symptoms includes the following:
-
Headaches
-
Facial flushing
-
If hypertension is severe, weakness, visual impairment, impaired consciousness, and seizures (hypertensive encephalopathy)
Hypokalemia can be precipitated by non–potassium-sparing diuretics or sodium loading. Symptoms of hypokalemia include the following:
-
Constipation
-
Polyuria and polydipsia (because of impaired renal concentrating ability)
-
Weakness
-
If the serum potassium is low enough, paralysis and disturbances of cardiac rhythm [3]
Workup in hyperaldosteronism
The aldosterone-to-renin ratio (ARR)—that is, the ratio of plasma aldosterone (expressed in ng/dL) to plasma renin activity (PRA, expressed in ng/mL/h)—is the most sensitive means of differentiating primary from secondary causes of hyperaldosteronism. It can be obtained under random conditions of sodium intake.
Tests for confirming autonomous aldosterone secretion include the following:
-
Saline infusion test
-
Oral salt loading test
-
Captopril test
-
Fludrocortisone suppression test
Tests for differentiating aldosterone-producing adenoma (APA) from other primary hyperaldosteronism include the following:
-
Postural testing
-
18-hydroxycorticosterone level
-
Dexamethasone suppression test
Adrenal venous sampling (AVS) is the criterion standard test to differentiate unilateral (APA or unilateral adrenal hyperplasia [UAH]) from bilateral disease in patients with primary aldosteronism (PA); however, it requires considerable skill.
Management of hyperaldosteronism
Surgical excision of the affected adrenal gland is recommended for all patients with hyperaldosteronism who have a proven APA. After surgical removal of an APA (aldosteronoma), a period of hypoadrenalism can occur. If this is not recognized, clinically significant hyponatremia and hyperkalemia may result.
Severe hypokalemia may require intravenous (IV) correction if the potassium concentration is less than 2.5 mmol/L or if the patient is clinically symptomatic. Once the potassium level is stable, sodium restriction and oral potassium supplements may be used as effectively as, or in addition to, potassium-sparing diuretics.
Spironolactone is the most effective drug for controlling the effects of hyperaldosteronism, though it may interfere with the progression of puberty. Newer drugs, such as eplerenone, that possess greater specificity for the mineralocorticoid receptor than spironolactone does are becoming available.
Idiopathic hyperaldosteronism
Control of hypokalemia and hypertension in idiopathic hyperaldosteronism (IHA) can be achieved with sodium restriction (to < 2 g/day) and the administration of spironolactone, eplerenone, or amiloride, but additional antihypertensives are often needed to achieve good control in this patient group.
Familial hyperaldosteronism type I (GRA)
In adult patients with familial hyperaldosteronism (FH) type 1 (FH-I), or glucocorticoid-remediable aldosteronism (GRA), control of hypertension can be achieved through treatment with physiologic doses of dexamethasone.
In children, however, dexamethasone is best avoided because of its adverse effects on growth and bone density. Hydrocortisone has a short half-life (a typical dose is 10-12 mg/m2) and is a better choice, but it is not as efficient at reducing mineralocorticoid levels. Amiloride may be a preferred option because it avoids the potential problems of growth retardation associated with the use of glucocorticoids and potential adverse effects resulting from the blockade of sex steroid receptors by spironolactone or eplerenone.
Familial hyperaldosteronism type II
Patients with FH-II should be regularly observed, and treatment should be started when they develop hypertension. Treatment is with the same agents as for IHA.
Familial hyperaldosteronism type III
The clinical spectrum of FH-III widely varies. Hence, some patients may benefit from medical treatment, whereas others require bilateral adrenalectomy due to resistance to aggressive antihypertensive therapy, including aldosterone receptor blockade and amiloride.
Pathophysiology
Normal aldosterone physiology
Aldosterone participates in the homeostasis of circulating blood volume and serum potassium concentration; these, in turn, feed back to regulate aldosterone secretion by the zona glomerulosa of the adrenal cortex. Aldosterone secretion is stimulated by an actual or apparent depletion in blood volume detected by stretch receptors and by an increase in serum potassium ion concentrations; it is suppressed by hypervolemia and hypokalemia.
The mechanisms regulating aldosterone secretion are complex, involving the zona glomerulosa of the adrenal glands, the juxtaglomerular apparatus in the kidneys, the cardiovascular system, the autonomic nervous system, the lungs, and the liver (see the image below). The major factors stimulating aldosterone production and release by the zona glomerulosa are angiotensin II and the serum potassium concentration. The juxtaglomerular apparatus is the principal site of regulation of angiotensin II production.
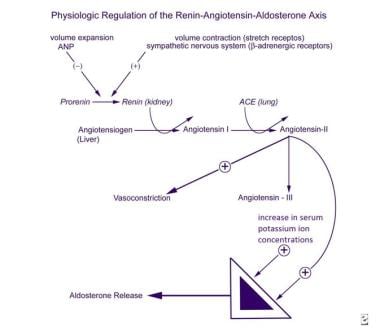
ACTH stimulates aldosterone secretion in an acute and transient fashion but does not appear to play a significant role in the long-term regulation of mineralocorticoid secretion. The major inhibitors of the zona glomerulosa include circulating atrial natriuretic peptide (ANP) and, locally, dopamine. Although ANP levels are clearly increased in hyperaldosteronism, neither ANP nor dopamine has been implicated as a primary cause of clinically disordered aldosterone secretion.
Metoclopramide has been shown to increase aldosterone secretion, suggesting that dopamine may tonically inhibit aldosterone release. The physiologic roles of adrenomedullin and vasoactive intestinal peptide (VIP) on aldosterone secretion remain to be clarified, although both of these neuropeptides are produced in rat zona glomerulosa.
The synthesis of prorenin, its conversion to renin, and its systemic secretion are stimulated by blood volume contraction detected by stretch receptors, beta-adrenergic stimulation of the sympathetic nervous system, and prostaglandins I2 and E2. These processes are inhibited by volume expansion and ANP.
Renin converts angiotensinogen, a proenzyme synthesized in the liver, into the decapeptide angiotensin I, which is then converted in the lungs into the octapeptide angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II is both a stimulator of aldosterone secretion and a potent vasopressor. Angiotensin II is metabolized to angiotensin III, a heptapeptide that is also a stimulator of aldosterone secretion.
The synthesis and secretion of prostaglandins I2 and E2 and the normal function of the stretch receptors are dependent on the intracellular ionized calcium concentration. Renal prostaglandin secretion is stimulated by catecholamines and angiotensin II. The complex regulation of aldosterone synthesis and secretion provides several points at which disturbance in the regulation of aldosterone secretion may occur.
Aldosterone is synthesized from cholesterol in a series of 6 biosynthetic steps (see the image below). Only the last 2 steps are specific to aldosterone synthesis; the first 4 also apply to cortisol synthesis by the zona fasciculata. Consequently, a defect in one of the specific aldosterone synthetic enzymes does not lead to hypercortisolism and secondary ACTH-mediated adrenal hyperplasia.
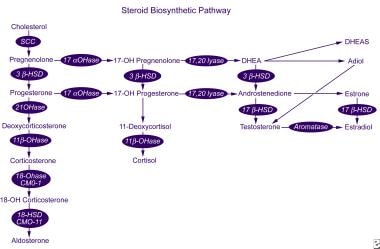
The enzyme aldosterone synthase is encoded by the gene CYP11B2 and has 11β-hydroxylase, 18-hydroxylase, and 18-hydroxydehydrogenase activity. This gene is located on human chromosome arm 8q24.3-tel, close to the gene CYP11B1, which encodes 11β-hydroxylase, the enzyme that catalyzes the final step of cortisol synthesis. Mutations in these genes can result in a number of disorders of aldosterone synthesis (see Differentials). [4]
Aldosterone action on target tissues (eg, the distal renal tubule, sweat glands, salivary glands, and epithelium of the large intestine) is mediated via a specific mineralocorticoid receptor. Mineralocorticoid receptors exhibit equal affinity for mineralocorticoids and cortisol, yet the aldosterone receptors in the distal tubule and elsewhere are protected from cortisol-mediated activation by 11β-hydroxysteroid dehydrogenase type 2, which locally converts cortisol to inactive cortisone.
Primary aldosteronism
The term primary hyperaldosteronism (or primary aldosteronism [PA]) refers to a renin-independent increase in the secretion of aldosterone. This condition is principally a disease of adulthood, with its peak incidence in the fourth to sixth decades of life.
More than 90% of cases of PA are due either to an aldosterone-producing adenoma (APA), which accounts for around 35% of cases (30-40%), or to idiopathic hyperaldosteronism (IHA), which accounts for around 60% of cases (almost all of which are bilateral). Unilateral adrenal hyperplasia (UAH) is a rare cause of PA, accounting for 1-2% of cases. About 1% of patients present with adrenocortical carcinomas that are purely aldosterone-secreting and are usually large at the time of diagnosis; 1% present with familial hyperaldosteronism, and 1% present with an ectopic aldosterone-producing adenoma or carcinoma. [5]
Unilateral adrenal hyperplasia accounts for 14-17% of all cases of unilateral PA. The prevalence of cortical adenoma within cortical hyperplasia is estimated to be 6-24%. The clinical presentation and outcome of patients with unilateral primary hyperaldosteronism are similar regardless of the histopathologic diagnosis. Unilateral adrenocortical hyperplasia is rare. [6]
APAs (sometimes referred to as aldosteronomas) are usually benign encapsulated adenomas that are less than 2 cm in diameter. Most cases are solitary, although in as many as one third of cases, evidence exists of nodularity in the same adrenal gland, suggesting that the condition has arisen in a previously hyperplastic gland.
Patients with IHA have bilateral thickening and variable nodularity of their adrenal cortex. A wide spectrum of severity exists for this disorder, which may go undetected for long periods with no hypokalemia and only mild hypertension. It has been suggested that IHA arises as a result of an undetected adrenal cortex–stimulating factor. Alternatively, the disorder may arise as a result of an activating mutation in an adrenal cortex–specific gene. Neither hypothesis has been proven.
Inherited forms of primary hyperaldosteronism account for only 1% of cases but are more likely to occur during childhood years. These forms include familial hyperaldosteronism (FH) types I, II, and III.
Familial hyperaldosteronism type I
FH type I (FH-I), also referred to as glucocorticoid-remediable aldosteronism (GRA), may be detected in asymptomatic individuals during screening of the offspring of affected individuals, or patients may present in infancy with hypertension, weakness, and failure to thrive due to hypokalemia. FH-I is inherited in an autosomal dominant manner and has a low frequency of new mutations.
The first clinical description of GRA appeared in 1966, and the genetic mechanism was discovered in 1992. FH-I arises as a result of unequal crossing over of highly related CYP11B1 (the 11β-hydroxylase gene) and CYP11B2 (the aldosterone synthase gene) during meiosis, producing an anti-Lepore-type fusion product. [7, 8] This genetic rearrangement causes the expression of CYP11B2 to be placed under the control of the CYP11B1 promoter and the aldosterone synthesis to be abnormally regulated by ACTH rather than by the renin-angiotensin system.
The result is ACTH-dependent aldosterone production and production of 17-hydroxylated analogues of 18-hydroxycortisol under ACTH regulation from ectopic enzyme expression in the zona fasciculata. Bilateral hyperplasia of the zona fasciculata occurs, and high levels of novel 18-hydroxysteroids appear in the urine. Adenoma formation is rare, but patients do have a significant increase in incidence of cerebrovascular aneurysms, for which they require screening.
Familial hyperaldosteronism type II
FH type II (FH-II) is a non–glucocorticoid-suppressible inherited form of hyperaldosteronism that was first recognized as a distinct entity by Gordon et al, though cases had previously been described in the 1980s. Like FH-I, it is inherited in an autosomal dominant manner. In contrast to FH-I, some FH-II kindreds exhibit a high rate of adenoma formation.
The mechanism and gene locus have not yet been identified, though CYP11B and the renin and angiotensin II receptor genes have been excluded. However, linkage has been established for a number of families to band 7p22. [9, 10] It has also been speculated that FH-II is not a single disorder.
Familial hyperaldosteronism type III
FH-III is a rare autosomal dominant form of PA characterized by early-onset hypertension, nonglucocorticoid-remediable hyperaldosteronism, and hypokalemia. Germline heterozygous missense mutations of the KCNJ5 gene, encoding Kir3.4, a member of the inwardly rectifying K+ channel family, have been identified as a cause of FH-III. Thus far, 4 mutations (G151R, G151E, T158A, and I157S) have been reported in 6 families. [11, 12, 13]
The clinical phenotype of patients harboring the above mutations ranges from severe PA and hypertension refractory to medical treatment that requires bilateral adrenalectomy, to mild or moderate hypertension responsive to medical therapy. In some patients, adrenal hyperplasia has been described.
Various studies from different centers report a prevalence of somatic KCNJ5 mutations in sporadic APAs ranging from 30-65%. [11, 14, 15, 16] There are 2 recurrent mutations, G151R and L168R, reported by all studies, whereas there is one report of a 3-nucleotide deletion, the delI157. [17]
The affected residues of both the germline and the somatic mutations are in or near the selectivity filter of the Kir3.4 potassium channel and are highly conserved among different species. Electrophysiologic studies demonstrate that these mutations result in loss of channel selectivity, with increased Na+ conductance leading to membrane depolarization. In zona glomerulosa cells, membrane depolarization leads to opening of voltage activated Ca2+ channels, with activation of the calcium-signalling pathway, the major mediator of aldosterone production.
APAs with KCNJ5 mutations are more prevalent in females than males and in younger patients. They are also associated with higher preoperative aldosterone levels. They are not related with the tumor size, but they are related with higher aldosterone levels and lower K+ concentrations.
Transcriptome and real-time polymerase chain reaction (PCR) analyses demonstrate that APAs with KCNJ5 mutations exhibit increased expression of the CYP11B2 gene and its transcriptional regulator NR4A2, thus increasing aldosterone production. It has also been found that APAs with and without KCNJ5 mutations display slightly different gene expression patterns. [16] Another study reports KCNJ5 mRNA levels higher in the APAs with KCNJ5 mutations and significantly higher in APA than cortisol-producing adenomas and pheochromocytomas. [15]
Somatic mutations in ATP1A1 (gene that encodes the alpha-1 [catalytic] subunit of the Na+/K+ ATPase, a member of the P-type ATPase family), ATP2B3 (gene that encodes the plasma membrane calcium transporting ATPase 3 [PMAC3], another member of the P-type ATPase family), or CACNA1D (gene that encodes Cav1.3, the alpha subunit of an L-type voltage-gated calcium channel) are present in approximately 6%, 1% and 8% of all cases of an aldosterone-producing adenoma, respectively. More recently, de novo germline mutations in CACNA1D were reported in 2 children with a previously undescribed syndrome that featured PA and neuromuscular abnormalities. [18]
Secondary hyperaldosteronism
Secondary hyperaldosteronism is a collective term for a diverse group of disorders characterized by physiologic activation of the renin-angiotensin-aldosterone (R-A-A) axis as a homeostatic mechanism designed to maintain serum electrolyte concentrations or fluid volume. In the presence of normal renal function, it may lead to hypokalemia.
Secondary hyperaldosteronism can be divided into 2 categories, 1 with associated hypertension and 1 without. The former category includes renovascular hypertension, which results from renal ischemia and hypoperfusion leading to activation of the R-A-A axis. The most common causes of renal artery stenosis in children are fibromuscular hyperplasia and neurofibromatosis. Hypokalemia may occur in as many as 20% of patients.
Plasma renin activity (PRA) levels are often in the reference range, but elevated levels of PRA may be detected after provocation with a single dose of captopril 1 mg/kg. Renal ischemia is also thought to underlie the secondary hyperaldosteronism observed in malignant hypertension.
Hyperreninemia and secondary aldosteronism have also been reported in patients with pheochromocytoma, apparently as a result of functional renal artery stenosis. Renin-producing tumors are very rare, and very high levels of PRA (up to 50 ng/mL/h) are noted, frequently with an increased prorenin-to-renin ratio. The tumors are generally of renal origin and include Wilms tumors and renal cell carcinomas.
Hyperkalemia due to chronic renal failure also causes secondary hyperaldosteronism. Low sodium-to-potassium ratios can be measured in saliva and stool. Cyclosporine-induced hypertension in solid organ transplant patients may also involve a component of hyperaldosteronism.
Secondary hyperaldosteronism in the absence of hypertension occurs as a result of homeostatic attempts to maintain the sodium concentration or circulatory volume or to reduce the potassium concentration. Clinical conditions in which it may arise include diarrhea, excessive sweating, low cardiac output states, and hypoalbuminemia due to liver or renal disease or nephrotic syndrome. Secondary hyperaldosteronism may also occur developmentally in newborn infants (see below).
Increased mineralocorticoid dependency in the young
The mineralocorticoid dependency of sodium reabsorption is increased during infancy and childhood, peaking in the neonatal period before decreasing progressively with advancing age. This increase occurs because the reabsorption of sodium and water by the proximal tubule is least efficient in early life, resulting in an increased sodium and water load at the level of the distal renal tubule.
Because sodium and water resorption from the distal tubule is mediated by the R-A-A axis, the PRA is approximately 10-fold to 20-fold higher in a newborn infant than in an adult. Consequently, neonates show relative increases in aldosterone production rates (>300 µg/m2/day vs 50 µg/m2/day in an adult) and plasma aldosterone concentrations (80 pg/dL vs 16 pg/dL). These increases in early life explain why young infants exhibit profound clinical symptoms of hyperaldosteronism that gradually improve with advancing age.
Etiology
The following is a summary of etiologies of hyperaldosteronism and conditions that mimic hyperaldosteronism:
Causes of primary hyperaldosteronism include the following:
-
APA - High aldosterone, low PRA
-
IHA - Responds to posture (bilateral adrenal hyperplasia)
-
Primary adrenal hyperplasia - Responds to posture (unilateral disease)
-
Chronic stress-induced hyperaldosteronism [19]
-
FH-I (GRA) - Sustained suppression of aldosterone (< 4 ng/dL) with dexamethasone
-
FH-II/FH-III - Familial (probably autosomal dominant)
Causes of secondary hyperaldosteronism include the following:
-
Edema disorders (eg, cardiac failure and nephrotic syndrome) - High aldosterone, nonsuppressed PRA (>2 ng/mL)
-
Renovascular hypertension
-
Renin-producing tumors
-
Pregnancy [20]
Causes of conditions that mimic aldosterone excess include the following:
-
Congenital adrenal hyperplasia (11β-hydroxylase deficiency and 17α-hydroxylase deficiency) - Low aldosterone, low PRA, elevated steroid intermediates
-
Primary glucocorticoid resistance - High glucocorticoid secretion unsuppressed by dexamethasone
-
Deoxycorticosterone-secreting tumors - Elevated deoxycorticosterone levels
-
Syndrome of apparent mineralocorticoid excess
-
Liddle syndrome
-
Gain of function mutation of mineralocorticoid receptor [21]
-
Licorice ingestion
-
Carbenoxolone
Hypokalemia may be precipitated by a diet that is rich in sodium or the concomitant administration of drugs that produce kaliuresis (including diuretics and carbenoxolone). Taking carbenoxolone or eating large quantities of licorice may result in hypokalemia because of the blockade of the target tissue enzyme that protects the aldosterone receptor from the relatively higher levels of circulating cortisol (apparent mineralocorticoid excess).
Epidemiology
Primary hyperaldosteronism is a rare condition in children. The youngest child reported with an aldosterone-secreting adenoma was aged 3 years. Earlier use of hypokalemia as a diagnostic requirement, as advocated by some authorities, may have led to underrecognition of the contribution of primary hyperaldosteronism to hypertension.
The prevalence rate for PA in hypertensive patients varies between studies, ranging from 4.6% to 16.6% in reports using confirmatory tests to diagnose PA. [22] Patients with PA also make up 17-23% of the treatment-resistant hypertensive population. [23, 24, 25, 26, 27]
Most of the hyperaldosteronism observed in the general population is sporadic, with most cases due to bilateral adrenal hyperplasia. APAs are likely to be diagnosed earlier than IHA because they are more likely than IHA to produce early symptomatic hypertension and hypokalemia. APAs account for 40% of cases of primary hyperaldosteronism.
It is possible that the distinction between adenoma and hyperplasia is not as clear as was once assumed. In one third of cases, associated hyperplasia or nodules of the adjacent zona glomerulosa is present, implying that the adenoma may have arisen in previously hyperplastic tissue.
Inherited forms of primary hyperaldosteronism (ie, FH-I [GRA], FH-II, and a very rare form known as FH type III [FH-III]) account for approximately 1% of cases of primary hyperaldosteronism, though they are more likely to occur during childhood and adolescent years than other forms of primary hyperaldosteronism are.
Studies of secondary hyperaldosteronism have found that approximately 15% of adults who attend hypertension clinics have elevated PRA. Reliable figures for children are not readily available.
Age-, sex-, and race-related demographics
Because the 2 causes that account for about 99% of cases of primary hyperaldosteronism have a peak age of onset in adulthood, the less common causes account for a larger percentage of children with hyperaldosteronism. For this reason, children with apparent hyperaldosteronism should be evaluated for evidence of congenital defects of the R-A-A axis and inherited forms of hypermineralocorticoidism.
Data on adults suggest that hyperaldosteronism has a female preponderance. Equivalent information is not available for children, in whom primary hyperaldosteronism due to inherited syndromes is likely to represent a greater proportion of cases.
The literature on adults demonstrates that blacks are at significantly greater risk for hypertension-related morbidity and mortality than whites are. They are also more likely to develop low-renin hypertension, though no studies indicate that the prevalence of primary hyperaldosteronism is significantly higher in blacks.
Prognosis
The age of the patient and the duration of disease before diagnosis are the 2 most important prognostic factors. Adult studies have shown that hypertension is cured in 30-60% of cases and significantly improved in 40-70% of cases, postoperatively (see Treatment). [5, 28] This figure is likely to be higher in children because disease duration is shorter and the prevalence of other causes of hypertension is lower.
Primary hyperaldosteronism can result in substantial morbidity and mortality as a result of hypertensive vascular complications (hypertrophy followed by sclerosis of intimal smooth muscle), renal complications (sclerosis), and cardiac complications (hypertrophy followed by dilatation). Through early recognition and treatment of hypertension, these complications can be avoided in children.
A study by Lai et al indicated that patients with autosomal dominant polycystic kidney disease (ADPKD) have a high prevalence of PA and that ADPKD patients with PA have an increased overall risk of cardiovascular disease. Surrogate markers showed a greater indication of atherosclerosis in these patients than in ADPKD patients with normal plasma aldosterone levels. The study included 27 hypertensive ADPKD patients, nine of whom (33%) had PA. [29]
Appropriate medical or surgical intervention in PA results in long-term reduction in blood pressure and left ventricle (LV) mass via LV inward remodeling (eg, through a reduction in LV diameters and volume). [30] Moreover, a significant decrease in urinary albumin excretion at 6 months after treatment has been reported in patients with PA and associated microalbuminuria. Both adrenalectomy and mineralocorticoid receptor antagonists (MRAs) can reverse the intrarenal hemodynamic pattern that leads to the decline in glomerular filtration rate and increased proteinuria. [13, 31] Furthermore, surgical or medical management of PA results in improvement in the metabolic complications of PA, such as plasma glucose control, and quality of life as well.
A study by Rossi et al found that patients with PA caused by an aldosterone-producing adenoma had, following adrenalectomy, a long-term atrial fibrillation–free survival rate comparable to that of optimally treated primary hypertension patients. However, medically treated patients with IHA had a lower long-term rate. The study’s median follow-up period was 11.8 years. The report’s results, according to the investigators, demonstrate the importance that early identification of PA patients requiring adrenalectomy has in the prevention of incident atrial fibrillation. [32]
The Aldosteronoma Resolution Score (ARS) is currently the most accurate prediction model for complete resolution of hypertension after adrenalectomy, taking into account 4 preoperative clinical parameters: body mass index (BMI) of 25 kg/m2 or higher, female sex, duration of preoperative hypertension 6 years or longer, and number of preoperative antihypertensive medications (≤2). Each parameter receives a score of 1, with the exception of number of preoperative medications, which is scored by 2 points due to its relative significance in the prediction model. A score of 0-1 predicts a low likelihood of resolution, whereas patients with ARS scores of 4-5 have a high likelihood of resolution of hypertension after adrenalectomy.
Moreover, data suggest that the TT genotype of the CYP11B2 gene encoding aldosterone synthase predicts resolution of hypertension in patients undergoing adrenalectomy for aldosterone-producing adenoma.
Patients with GRA must undergo assessment of their cerebral circulation because this disorder is associated with a significant risk of cerebral vascular aneurysms. Provided that hypertension is well treated, morbidity and mortality should not be increased significantly.
Hypokalemia is more frequently observed in patients with adenomas, though it should not be considered a diagnostic feature of primary hyperaldosteronism, as was once thought. Patients with adenomas are more likely to develop this complication, as are patients who have milder disease but receive treatment with diuretics for their hypertension before the hyperaldosteronism is diagnosed.
Hypokalemic patients may experience neuromuscular symptoms such as weakness or paralysis, constipation, and polyuria and polydipsia because of an associated renal concentrating defect. Hypokalemia also impairs insulin secretion and can promote the development of diabetes mellitus.
Although cardiac fibrosis has been reported in adults with primary hyperaldosteronism, no such reports exist in children, possibly because of the shorter duration of disease at the time of diagnosis. Cardiac fibrosis has also been reported in rats treated with excessive amounts of mineralocorticoids, especially if hyperglycemia is also present. This effect can be ameliorated with amiloride. The role of aldosterone in diabetic heart disease has been questioned, and trials of mineralocorticoid antagonists in this condition have been initiated.
Patient Education
Patients with mild hyperaldosteronism must learn how to avoid foods that are high in sodium; such foods will exacerbate their hypertension and increase their tendency to develop hypokalemia.
Patients also should be informed that medications can lead to hyperkalemia and hypotension, particularly in the presence of intercurrent illness, and should be advised to see their pediatrician if these conditions develop.
-
Steroid biosynthetic pathway.
-
Physiologic regulation of the renin-angiotensin-aldosterone axis.
Tables
False Negative Results |
|||
Factor |
Aldosterone |
Renin |
ARR |
Medications |
|
|
|
K-sparing diuretics |
↑ |
↑↑ |
↓ |
K-wasting diuretics (Non-K-sparing diuretics, such as thiazides, induce renal potassium losses and reduce plasma potassium concentrations, leading to decreased aldosterone secretion.) |
→↑ |
↑↑ |
↓ |
ACE inhibitors |
↓ |
↑↑ |
↓ |
Angiotensin receptor blockers |
↓ |
↑↑ |
↓ |
DHPs (It is a shared opinion that dihydropyridinic calcium channel blockers do not significantly affect aldosterone secretion, mainly causing an increase in PRA, which rarely gives false negatives.) |
→↓ |
↑ |
↓ |
Other conditions |
|||
Hypokalemia |
↓ |
→↑ |
↓ |
Sodium-restricted diet |
↑ |
↑↑ |
↓ |
Pregnancy |
↑ |
↑↑ |
↓ |
Renovascular hypertension |
↑ |
↑↑ |
↓ |
Malignant hypertension |
↑ |
↑↑ |
↓ |
False Positive Results |
|||
Beta-adrenergic blockers |
↓ |
↓↓ |
↑ |
Central alpha-2 agonists (eg, clonidine, alpha-methyldopa) |
↓ |
↓↓ |
↑ |
NSAIDS |
↑ |
↓↓ |
↑ |
Other conditions |
|||
Potassium loading |
↑ |
→↓ |
↑ |
Sodium-loaded diet |
↓ |
↓↓ |
↑ |
Advancing age |
↓ |
↓↓ |
↑ |
Renal dysfunction |
→↑ |
↓ |
↑ |
PHA-2 |
→ |
↓ |
↑ |
Luteal phase of menstrual cycle |
↑ |
PRA: Unchanged |
↑ |
Antihypertensive Medications With Minimal Effect on the ARR |
|||
Prazosin, doxazosin, terazosin |
|
←→ |
|
Verapamil, hydralazine |
|
←→ |
|
Other medications |
|||
Renin inhibitors (Renin inhibitors raise the ARR if renin is measured as PRA [false positive] and lower it if measured as DAR concentration [false negative.]) |
↓ |
↑↓ |
↑↓ |
SSRIs |
↑ |
↑ |
↓ |
OCPs (OCPs have little effect on ARR when renin is measured as PRA. Use of immunometric measurements of DAR rather than PRA may give false positive results. Subdermal etonogestrel has no effect on ARR.) |
↑ |
↓DAR |
↑ |
Liddle syndrome |
↓ |
↓ |
Normal |
ARR, aldosterone-renin ratio; NSAIDs, non-steroidal anti-inflammatory drugs; K, potassium; ACE, angiotensin converting enzyme; ARBs, angiotensin II type 1 receptor blockers; DHPs, dihydropyridines; PHA-2, pseudohypoaldosteronism type 2; PRA, plasma renin activity; DAR, direct active renin; OCPs, oral contraceptive agents; SSRIs, selective serotonin reuptake inhibitors |
|||
Drug |
Class |
Pediatric Dose |
Spironolactone |
Aldosterone antagonist |
0-10 kg: 6.25 mg/dose PO q12h 11-20 kg: 12.5 mg/dose PO q12h 21-40 kg: 25 mg/dose PO q12h >40 kg: 25 mg PO q8h |
Potassium canrenoate |
Aldosterone antagonist |
3-8 mg/kg IV qd; not to exceed 400 mg |
Amiloride |
Potassium-sparing diuretic |
0.2 mg/kg q12h |
Triamterene |
Potassium-sparing diuretic |
2 mg/kg/dose q8-24h |
Nifedipine |
Dihydropyridine calcium channel antagonist |
0.25-0.5 mg/kg PO q6-8h |
Amlodipine |
Calcium channel antagonist |
0.05-0.2 mg/ day PO |
Doxazosin |
Alpha1 -specific adrenergic antagonist |
0.02-0.1 mg/day; not to exceed 4 mg |
Prazosin |
Alpha1 -specific adrenergic antagonist |
0.005 mg/kg test dose, then 0.025-0.1 mg/kg/dose q6h; not to exceed 0.5 mg/dose |







